水质的检验
出处:按学科分类—工业技术 中国轻工业出版社《焙烤工业实用手册》第719页(4332字)
(一)水样的采集
一般理化分析需水样2L,可用无色磨口具塞的硬质玻璃瓶或聚乙烯塑料瓶装。采样时先用水样冲洗装样瓶3次,然后按表4-2-31方法取水。
表4-2-31 取水样的方法

取样完毕后,塞好瓶塞,用塑料薄膜包扎瓶口(如长途运送,最好用石蜡封严),贴好标签,注明采样的时间、地点、来源及周围环境等。
采样后应马上分析,否则应放阴凉处或冰箱的冷藏室,供理化分析的水样允许存放的时间为:泉水和没有污染的井水为72h;清洁的河水和其他稍受污染的水为48h;受污染的水为12h。
(二)水质的理化检验
1.pH的测定
(1)仪器:
①pH计(酸度计):pHS-2或25型。
②玻璃电极:231型或221型。
③甘汞电极:232型或222型。
④塑料杯:100mL。
(2)试剂:标准缓冲溶液为pH4.00和pH9.20配制:25℃时量取0.1000mol/L氢氧化钠0.40mL和0.20mol/L邻苯二甲酸氢钾25.00mL,混合为pH4.00的标准缓冲溶液。
量取0.1000mol/L氢氧化钠26.70mL和0.2mol/L硼酸氯化钾25.00mL,混合为pH9.20标准缓冲溶液。
(3)操作:
①按照酸度计说明书所示的操作方法进行。
②将电极和塑料杯用水冲洗洁净后,用标准缓冲溶液冲洗1~2次。
③把标准缓冲溶液放入塑料杯中(约70mL),插入电极,校正仪器刻度。
④用水样将电极和塑料杯冲洗6~8次后,测水样的pH。
⑤测定后,将电极和塑料杯冲洗干净,妥善保存。
(4)注意事项:
①由于水样的pH受空气中CO2等因素影响而改变,因此采集水样后,立即测定,不宜久存。
②新的玻璃电极在使用前,必须在水中或0.1mol/L盐酸中浸泡一昼夜以上。不用时,也最好浸泡在水中。
在使用甘汞电极时,应把加氯化钾溶液处的橡皮塞拔下,使毛细管保持足够的液位差,从而有少量氯化钾溶液从毛细管中流出。否则,试液进入毛细管,将影响测定效果的准确度,不用时应将小橡皮塞塞上,再把电极下端的橡皮帽套上,不应浸泡在水中。
③测定时,应确保甘汞电极和玻璃电极之间形成回路,为此应检查甘汞电极毛细管是否畅通。检查方法是:先将毛细管擦干,然后用滤纸贴在毛细管的末端,如有溶液渗下,则证明毛细管未堵塞。同时尚需检查电极内溶液有无气泡隔断,液面是否低于内部电极。
④甘汞电极内溶液,应有少许氯化钾结晶存在,以证明确为饱和溶液。
⑤玻璃电极极易损坏,使用时应特别小心。如果玻璃膜沾有油污,可先浸入乙醇,然后浸入乙醚或四氯化碳中,最后,再浸入乙醇中,用水冲洗洁净。
2.总硬度的测定
(1)试剂:
①NH3H2O-NH4Cl缓冲溶液:溶16.9g氯化铵于143mL浓氨水中,用水稀释至250mL,贮于塑料瓶或硬质塑料瓶中,紧密塞好,严防氨水逸出或二氧化碳进入。若经常开启使用不得超过一个月。
②三乙醇胺:1∶1。
③盐酸羟胺溶液(1%):溶1g盐酸羟胺于水中,稀释至100mL。
④铬黑T指示剂:0.5g铬黑T溶于10mL缓冲溶液中,用95%乙醇稀释到100mL,贮于棕色瓶中,放在冰箱内保存,也可用固体铬黑T与氯化钠按1∶100混合研磨后长期使用。
⑤EDTA标准溶液(0.01mol/L):称3.723gEDTA,溶于水中,稀释至1000mL。吸取25mL用水稀释至50mL,然后用标准钙溶液标定,EDTA的量浓度计算如下:
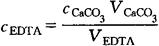
式中 VEDTA——EDTA溶液的用量(mL);
cCaCO3——标准钙溶液的量浓度(mol/L);
VCaCo3——标准钙溶液的用量(mL)。
标准钙溶液:称量1.000g无水碳酸钙(CaCO3)于500mL锥形瓶中,在瓶颈中放一漏斗,分次少量加入1∶1盐酸直到碳酸钙完全溶解,加200mL水,煮沸数分钟,以除去二氧化碳。冷却后加几滴甲基红指示剂,并加适量3mol/LNH3H2O或1∶1盐酸调节至橙色,定量转移至1L容量瓶中,用水定容,此溶液的浓度为0.01mol/L。
(2)操作:取水样25.00mL或更适当体积(以滴定时消耗EDTA标准液不少于15mL,从加入缓冲液至滴定结束,时间不超过5min为宜)于250mL锥形瓶中,用水稀释至50mL,加1~2mL缓冲溶液,1~2滴铬黑T指示剂溶液或固体粉末,慢慢滴入EDTA标准溶液,并充分摇动,直至溶液由葡萄酒红色经紫蓝色变成蓝色,即为终点。在滴定最后几滴时,两滴之间应间隔3~5s。
(3)计算:水的硬度以mg/kg计,即100万份水样中含CaCO3的份数也相当于1L水中含CaCO31mg,故得:

式中 mCaCo3——碳酸钙的质量(mg);
CEDTA——EDTA标准溶液的量浓度(mol/L);
VFnTA——为滴定时用去的EDTA标准溶液的体积(mL);
MCaCo3——单位CaCO3量所具有的质量(g/mol)。
(4)说明:
①在滴定时,若终点不清楚,如不是指示剂变质,则可在水样中加入缓冲溶液后,加入适当的掩蔽剂,再加指示剂重新滴定。对铜的干扰,加0.5~4.5mL硫化钠溶液;对锰的干扰,加0.2~2mL盐酸羟胺溶液,或在指示剂中加入适当盐酸羟胺;对少量锰、铜、铝的干扰,也可加1~3mL三乙醇胺消除。若在滴定前加热水样至30~40℃或适当稀释也可以使终点更清楚。
②为了使滴定终点敏锐,在配制缓冲溶液时,可加入1.25gEDTA的镁盐,在缓冲溶液稀释前加入,也可用0.644g水合氯化镁(MgCl2·6H2O)溶于50mL水中代替。
3.铁的测定
(1)试剂:
①亚铁标准溶液:称取0.7020g硫酸亚铁铵晶体[Fe(NH4)(SO4)2·6H2O],溶于50mL蒸馏水中,加入20mL浓硫酸,再用蒸馏水稀释到1000mL。此溶液1.00mL含0.100mg亚铁,再用移液管吸取此溶液10.0mL,加蒸馏水稀释至100mL,此溶液1.00mL含10μg亚铁。
②10%盐酸羟胺溶液(现配现用)。
0.1%邻菲罗啉溶液。
(2)测定:
标准曲线的绘制:取50mL容量瓶6只,分别准确吸取10μg/mL铁标准溶液1.0、2.0、4.0、6.0、8.0、10.0mL于各容量瓶中,各加1mL10%盐酸羟胺溶液,摇匀,经2min后再加5mL1molNaAc溶液及3mL0.1%邻菲罗啉溶液,以水稀释至刻度,摇匀。在721型分光光度计上,用2cm厚比色皿,在最大吸收波长(510nm)处,测定各溶液的吸光度。以铁含量为横坐标,吸光度为纵坐标,绘制标准曲线。
取水样1~10mL(视水中含铁量而定),代替标准溶液,其他步骤均同上,测其吸光度。根据未知液的吸光度,在标准曲线上查出未知液中的铁含量,并以每毫升未知液中含铁微克数表示。
(3)计算:

式中 V——取水样的毫升数(mL);
m——标准曲线上查得铁的微克数(μg)。
4.铅、砷的测定
参见第二章第二节。
(三)微生物检验
参见第二章第三节。