遗传性肾炎
出处:按学科分类—医药、卫生 科学技术文献出版社《肾脏内科疾病诊断标准》第309页(6738字)
一、概述
遗传性肾炎(hereditary merulonephritis),又称Alport综合征(alport syndrome,AS)、眼—耳—肾综合征,是以血尿、进行性肾功能不全、伴或不伴感音性耳聋和眼部改变为临床特征的遗传性肾小球基底膜(glomerular basement membrane,GBM)疾病。男女均可发病,但重症者多为男性。1902年Guthrie首先报道该病,20世纪20年代Alport命名了本病,20世纪70年代发现了本病的GBM具有典型的超微病理改变,20世纪80年代揭示了本病的GBM中正常Ⅳ型胶原成分异常。20世纪90年代,有关本病的致病基因被定位和克隆,在大量患者中检测到基因突变,对本病发病机理的认识取得了突破性的进展。
二、流行病学
以往认为AS是一种罕见的遗传性疾病,随着对其研究的深入和诊断水平的提高,目前认为此病并不罕见。据文献报道,AS发病率为1∶(5000~10000),在肾小球疾病中约占2%,在小儿慢性肾功能不全病例中约占3%,在肾移植病例中约占2.3%,在终末期肾衰患者中约占5%,在成人肾活检中占0.3%,而在儿童肾活检中占1.7%~2.5%。
三、病因及发病机理
近年研究发现,本病的发生与基底膜Ⅳ型胶原α链编码基因突变有关。Ⅳ型胶原纤维主要分布在基底膜,是肾小球滤过膜的重要结构基础。它含有6条α链,分别由基因COL4A1、COL4A2、COL4A3、COL4A4、COL4A5和COL4A6编码。Ⅳ型胶原纤维为三螺旋结构,是由3条α链即α1、1、2(Ⅳ),α3、4、5(Ⅳ),α5、5、6(Ⅳ)组成,这3条α链无论是在碱基长度,还是在甘氨酸重复序列的长度与所在位置上都具有很强的相似性。因此其中任何一条α链上发生突变,即可引起其构象的改变,从而导致整个三螺旋结构稳定性的丧失,或形成不能正常沉积于基底膜上的异常Ⅳ型胶原分子。Ⅳ型胶原分子结构和功能的异常影响基底膜的完整性,从而出现相应的临床表现。近年应用分子生物学技术对本病进行研究,其遗传方式、基因部位及基因产物的异常,见表7—1。
表7—1 AS与各链编码基因关系
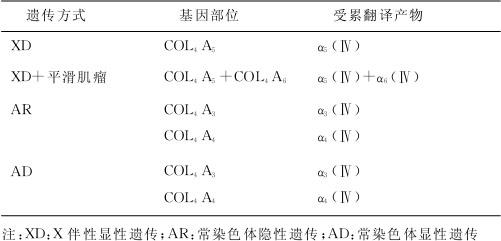
有研究表明,除了Ⅳ型胶原基因编码的遗传分子缺陷,主要组织相容性等位基因也可能与这些缺陷的胶原基因不均衡有关。还有研究发现,AS患者在基因上有同样的突变,而其临床表现却大不相同,说明其他基因或环境也影响着疾病的发生发展。
四、遗传方式
本病遗传呈异质性,遗传方式包括X伴性显性遗传(XD)、常染色体显性(AD)及常染色体隐性遗传(AR)。
1.XD 最为常见,占80%~85%,XD的致病基因位于X染色体长臂中部Xq21—q22上,为COL4A5和COL4A6,即编码基底膜主要组成成分—Ⅳ型胶原α5、6链的基因。在这类家系中,父病只传女不传男,母病传女也传男,因此家系女性患者多于男性,而男性病情明显比女性重。研究认为,XD男性与女性基底膜的Ⅳ型胶原组成不同导致临床表现不同,男性α5链染色阴性,由于α5链的结构异常,不能形成正常的α3、α4、α5三螺旋结构并易于降解,故α3、α4链在基底膜中的染色也是阴性的,而XD女性基底膜染色是间断阳性的。
2.AR 约占10%,部分家系以此方式遗传,其致病基因定位于第2对常染色体长臂远段2q35—q37上,为编码Ⅳ型胶原α3、4链的基因COL4A3和COL4A4,以COL4A3基因突变居多。由于致病基因在常染色体上,故遗传与性别无关,以此方式遗传的家系中男女得病机会均等,患者疾病严重程度也与性别无关。
3.AD 此方式遗传最为少见,1981年后AS才有此遗传方式的报道。其发病可能与COL4A3和COL4A4基因有关。该致病基因虽也在常染色体上,但杂合子的表现型正常,惟纯合子才显病,故具有临床症状的患者常为近亲婚配的子女。该致病基因在染色体上的定位尚未见报道。
绝大部分AS患者以上述3种遗传方式遗传,但有约5%患者为基因新突变所致,无家族史。
五、病理
1.光镜 无特异性。疾病早期肾小球可基本正常或病变很轻,随疾病的进展可发展为局灶节段硬化或弥漫系膜细胞增生、基质增多,渐发展至肾小球硬化,少数有新月体形成,肾间质可从炎症细胞浸润发展到纤维化,并伴肾小球不同程度的萎缩。部分患者有大量间质泡沫细胞,常首先出现于皮、髓质交界处,但它并不是本病特有,也可以见于肾病综合征、肾小球肾炎及肾盂肾炎,不过仍以Alport综合征最常见,阳性率约40%,因此对提示本病仍有一定意义。另外,10%~25%的Alport综合征患者具有胎儿型肾小球。
2.免疫荧光 多为阴性,少数标本可有Ig、补体的局灶沉积。
3.电镜 具有特征性,随着患者年龄增长疾病随之进展,GBM将逐渐出现异常。其主要病变有3种:GBM增厚并劈裂,呈板层样改变;GBM变薄;二者相间。三者可以同时交替出现,或者以某一种为主。现认为随病变进展,变薄的GBM可向增厚转换。增厚的GBM厚度可达正常的2~5倍,其上皮侧缘常呈不规则波浪形,增厚的致密带纵向劈裂分层,且相互交错成网,网眼中可见微小电子致密颗粒。若增厚的GBM广泛存在,且与变薄的GBM相间出现,对本病诊断极有意义。
六、临床表现
1.肾脏表现 血尿是最早、最常见的临床表现,几乎全部患者均有血尿,男性多表现为持续镜下血尿,女性多为间歇性镜下血尿,30%~70%患者可伴反复肉眼血尿,其发生常与上呼吸道感染有关。男性患者在疾病早期常不表现有蛋白尿,但随着疾病的进展,多数患者出现蛋白尿,且逐渐加重,约30%患者的蛋白尿甚至发展到肾病综合征水平。女性患者也可表现有显着的蛋白尿,但并不常见。肾功能往往呈进行性下降,特别是男性,大多在30岁之前进入终末期肾衰竭,而女性肾功能受累出现较晚。高血压亦是一常见症状。
2.听力损害 累及50%~70%患者,为双侧、对称性、感音性神经性耳聋,最常累及频率范围为2000~8000Hz,部分男性、病情严重患者可有其他频率范围的累及,表现为听力进行性减退,女性伴发耳聋者较少、出现亦较晚。多数患者耳聋程度与肾功能损伤程度相平行。
3.眼部改变 见于15%~30%的患者,眼虽可出现多种病变,但现认为只有前锥形晶体及黄斑周围视网膜黄白色颗粒沉积(点彩样改变)才为本病的特征表现,后者主要见于已发生肾功能损害的患者。其他有后锥形晶体、球形晶体等,可引起视力下降、弱视、眼球震颤,甚至晶体混浊。
4.平滑肌瘤 见于部分X伴性显性遗传患者,可见于食道、支气管、女性生殖系统的弥漫性平滑肌瘤病(DL),可表现吞咽困难、胸痛、反复支气管炎、咳嗽等症状。有学者认为,与DL相关的AS患者成人中发生肾衰竭可能会较晚。
5.其他 可并发肾盂肾炎、多发性周围神经病变、巨血小板减少性紫癜、肌肉发育不良、多发性畸形、高脯氨酸血症、甲状腺和甲状旁腺疾病等。
七、检查
1.一般检查 可有血尿(尿红细胞形态检查提示多数为肾小球性血尿,但有少数患者也可以为非肾小球性血尿)、蛋白尿、进行性血肌酐升高、贫血。
2.眼科检查 扩瞳后裂隙灯观察晶体形状,眼底镜观察眼底视网膜改变。
3.电测听检查 同时检查气导和骨导,刺激频率250~16000Hz。若气导和骨导平行性下降则为感音性神经性耳聋。
4.皮肤及肾组织Ⅳ型胶原α链间接免疫荧光检测 正常情况下,GBM、包氏囊、远端小管基底膜上有α3、4、5(Ⅳ)链沉积,表皮基底膜上有α5(Ⅳ)链的沉积,免疫荧光呈线形改变。在XD—AS中,男性肾组织上α3、4、5(Ⅳ)链免疫荧光均为阴性,皮肤α5(Ⅳ)链亦为阴性,女性则呈不连续的沉积;而在AR—AS中皮肤、肾小球包氏囊、肾小管基底膜α5(Ⅳ)链免疫荧光阳性,而α3、4、5(Ⅳ)链均为阴性。该检测方法具重要的诊断价值,并有助于遗传方式的确定,对于X伴性显性遗传患者,在丧失肾穿刺情况下,皮肤组织检测仍可帮助诊断。与电镜相比,具有操作简单、设备要求低等优点。
八、诊断
AS至今缺乏统一诊断标准,目前根据临床表现(肾、听力、眼等脏器病变)、家族史、病理改变、Ⅳ型胶原不同α链检测,结合以上4项,综合判断。
在肾脏超微结构改变中比较注重强调GBM增厚、致密层撕裂、分层、呈网状或编篮状等变化。但在实际工作中并非如此,往往这些特征性超微结构变化并不很明显,尤其在AS早期、儿童和成人女性患者常常很难发现上述典型的超微病理特征,而一些病例主要表现为GBM弥漫变薄或厚薄不均。因此临床和病理往往不能达成一致的诊断。青少年和女性常表现为GBM弥漫变薄,而老年患者和男性为则表现为GBM明显增厚和编篮状。一些学者认为如果增厚的GBM广泛存在并与变薄的GBM相间出现,对本病的诊断极有意义。
Ⅳ型胶原α链的检测有重要诊断价值。如果α5(Ⅳ)链染色阴性,可确诊X连锁型AS;但如α5(Ⅳ)链显示正常,尚不能断然除外AS诊断,还应考虑并除外如下可能:①非X连锁型AS,如AR—AS,α5(Ⅳ)在包氏囊及肾小管基底膜染色可阳性,其余肾组织Ⅳ型胶原α链染色均阴性;②少部分X连锁型AS患者中,4%~30%皮肤免疫荧光染色也可正常。
突变基因的检测是确诊的重要手段,尤其对于携带者检出、产前诊断,国内外均有开展AS相关基因突变检测,但由于无“热点突变”发现、突变形式多样、涉及多个基因和一个基因内几乎所有外显子及DNA分析技术限制等原因,目前仅限于实验室研究。
九、诊断标准
1.Flinter临床诊断标准(1988年)
(1)阳性家族史。
(2)耳病变:高频感音性耳聋。
(3)眼病变:前锥形晶体、黄斑周围黄白色颗粒。
(4)肾脏病变:血尿、进行性肾功能不全、肾小球基底膜典型超微结构改变。
以上4条具备3条可诊断AS,然而研究表明45%~55%的AS患者表现有耳聋,眼部异常的发生率仅为30%~40%,因此Flinter这一标准过于严格,如仅表现肾脏改变或无家族史者等,可致使许多患者漏诊。
2.Gregory提出的诊断标准(1996年)
(1)肾炎家族史。
(2)持续血尿,排除其他可能的遗传性肾脏病如薄基底膜病、多囊肾、或IgA肾病。
(3)双侧神经性耳聋,累及频率范围2000~8000Hz。听力呈进行性丧失,幼年无听力损害,而30岁之前已很常见。
(4)COL4An(n=3,4,5)基因突变。
(5)免疫组化检测发现肾小球和(或)表皮基底膜有Ⅳ型胶原不同α链全部或部分丧失。
(6)弥漫肾小球基底膜超微结构改变,尤其弥漫厚薄不均和断裂。
(7)眼病变包括前锥形晶体、后囊下白内障、视网膜病变等。
(8)一个家系中至少有2个家系成员逐渐进展至ESRD。
(9)巨血小板减少性紫癜或粒细胞性内涵物。
(10)弥漫食管和(或)女性生殖系统平滑肌瘤形成。
以上10条标准符和4条即可诊断。诊断AS家系,直系家庭成员必须符合4条标准(并不是同一例必须具备4条标准),当考虑旁系成员或仅表现为不明原因血尿、ESRD或听力障碍的极个别个体时应十分慎重;判断AS家系中家庭成员是否受累时,如果该个体符合相应遗传型,且符合标准2~10中的1项,可做拟诊,符合2项便可诊断;对于无家族史患者的诊断,至少应符合4条标准。
十、鉴别诊断
AS主要需与家族性IgA肾病、薄基底膜病、指甲—髌骨综合征等疾病进行鉴别,其临床表现比较相似,家族史、电镜下GBM改变和Ⅳ型胶原α链检测可有助鉴别。
1.家族性IgA肾病 肾脏表现与AS相似,但无眼、耳特征性改变,无Ⅳ型胶原异常,肾脏病理检查见免疫荧光表现以IgA为主的免疫球蛋白沉积,电镜见大块电子致密物沉积,无GBM分层、网状、断裂改变,可呈局灶厚薄不均。
2.薄基底膜病(TBMN) AS与TBMN的病变都与Ⅳ型胶原α链的编码基因突变有关,因此造成两者在临床表现、病理改变上有相似之处。尤其是部分早期AS患者仅表现为镜下血尿、肾功能正常,而无眼、耳体征,此时临床上很难与TBMN区别。该病超微结构主要表现为GBM弥漫性变薄,但不存在Alport综合征的肾小球基膜分层、增厚等特点。而年幼AS患者、任何年龄XD的女性及个别成年AS男性患者的GBM呈现弥漫性变薄时,需要与其进行鉴别诊断,部分薄基底膜病可能为不典型Alport综合征(见表7—2)。
表7—2 Alport综合征与薄基底膜病的鉴别


3.指甲—髌骨综合征 是一种罕见的遗传性疾病,临床以指甲和骨关节发育异常为主,表现为指甲发育不良或缺如,髌骨发育不全或缺失,髂骨后骨刺,桡骨头和肱骨小头发育不全而使桡骨头易半脱位,并可累及肾脏、眼等多脏器,需与AS鉴别。该病眼部病变包括色素性虹膜炎、青光眼、白内障、虹膜缺失、小角膜等;高频感音性耳聋,心、脑、甲状腺亦可有异常改变。30%~50%患者合并肾脏疾病,病程呈慢性进展,平均33岁左右进入ESRD。肾脏组织超微结构见GBM不规则增厚,增厚部位有散在致密胶原纤维沉积,呈“虫蚀”状改变,系膜区可有类似物质沉积为本病特征性改变。