功能性物质的变化
出处:按学科分类—医药、卫生 中山大学出版社《脊髓损伤》第541页(7796字)
脊髓损伤后发生的一系列病理生理反应,其所含的一些功能性物质会发生种类及数量的变化。下面重点讲述脊髓损伤后神经递质、NO以及自由基的变化。
一、神经递质的改变
脊髓急性损伤后,血供的改变可以引起一系列代谢方面的变化,后者又将影响损伤部位及其附近神经递质的调节和释出。Osterholm等发现脊髓损伤后,有大量的去甲肾上腺素聚集。该作者提出儿茶酚胺学说,认为脊髓内儿茶酚胺是引起血管变化的原因,而非外伤直接作用在血管的平滑肌,引起其收缩,使管腔变窄,阻力增加,血流变慢。他解释脊髓损伤后所以出现延迟出血的原因是由于神经递质需要一段时间聚集才能达到毒性浓度。Osterholm还通过实验在造成脊髓损伤前,如在损伤部位以上将脊髓横断或半横断,或将损伤部位邻近的几对神经根切断,都会大大降低脊髓出血性坏死的严重程度。这些结果表明,脊髓损伤后的组织改变波及后根及后柱部分,在脊髓损伤自体破坏过程中需要有完整的脊髓和神经根传入或传出通路的作用。Abraham等报告脊髓损伤后5-羟色胺(5-HT)增加。在正常稳定状态下,5-HT不越过血-脑屏障,但在脊髓损伤后的急性阶段,血-脑屏障受到破坏,5-HT可在单胺能神经末梢及内皮细胞的突触裂隙中被释放进入血流。在血小板膜上有特异性D-型5-HT受体。5-HT与这类受体结合可使血小板聚集。5-HT被认为是一种血管收缩剂,组织及脑脊液胺水平的增加可影响局部血流。5-羟吲哚乙酸(5-HIAA)的增加也能证明组织中5-HT的增加。可能增加的5-HT漏至局部血管引起血小板聚集而致使脊髓中央性出血性坏死。5-HT还可以引起自由基的形成,而使脂质过氧化增加。早在脊髓挫伤后半小时,就可观察到坏血酸引起的脂质过氧化增加,可能由于在所有损伤的细胞膜易发生过氧化的多不饱和的脂肪酸增加所致。损伤后由于出血,中央灰质血液的外渗可以引起自由基反应。出血后,铁复合物释放至组织可对膜脂开始催化自由基过氧化损害。脊髓挫伤后4小时,脂质过氧化只有不明显增加,而在压迫伤所有时间,这种增加均明显。压迫4小时后给予减压,可使坏血酸引起的脂质过氧化明显减少。Naftchi等对大鼠胸段脊髓横断后观察中枢神经系统及其他生命器官酪氨酸羟化酶、神经递质去甲肾上腺素(NE)、多巴胺(DA)、组织胺(HA)、cAMP的浓度变化。结果发现在损伤部位以上,NE较正常增加3倍,5-HT增加2倍。而在损伤部位以下明显降低,7~12天以后不能测出。在脑干,脊髓横断后7天,HA、NE和5-HT明显高于对照组;在脑,损伤后第一个24小时内,HA、NE和5-HT又急剧波动,以后5-HT及HA降至对照水平,而NE在伤后7天是明显下降,随后恢复正常水平。Snider等对大鼠颈髓横断,在伤后18~24小时,整个脑的神经递质水平及肾上腺儿茶酚胺发生改变。在肾上腺DA降低,而NE及E(肾上腺素)增加。在脑,DA及5-HT增加10%~15%,而GABA下降,这可能是由于神经冲动及血-脑屏障的改变及神经递质代谢所需的物质的有效性改变所致。切断这些下行单胺能抑制结构,将使从周围到运动神经元池的输入增加,而加强中枢神经系统的应急性。刘英炳等在狗的腰部用乳胶管制成的压迫囊注入盐水对脊髓压迫致伤,压迫1小时,然后以T7处脊髓作为对照,并同时取损伤上端、损伤段和损伤下端脊髓测定神经递质。实验结果表明,损伤上端和损伤下端5-HT明显升高。损伤段5-HIAA的含量显着下降,与损伤段5-HT含量呈显着正相关。Eghwrudjaktor等对大鼠T10髓节施以穿入伤,应用高效液相色谱-电化学探测仪(HPLC-ECD)发现损伤部位相邻近、远侧节段其单胺数值与对照组无损伤者无显着差异。在损伤段,伤后2小时所有生物胺浓度均明显下降,以后缓慢上升。伤后48小时,NE、DA、5-HT以及NE的代谢产物3-甲氯基-4羟基苯乙二醇、5-HT的代谢产物5-HIAA仍较对照组为低,但DA的代谢产物3,4-二氢苯乙酸及高香草酸稍增高。在伤后2小时,所有神经递质及其代谢均降低,说明其代谢及释放受到障碍。中枢神经系统对损伤的基本反应表现为因反射性痉挛所致的缺血,脑与脊髓对穿透伤生物胺的反应不同,说明除缺血外还可能有其他因素。Rawe等在损伤前给予甲基酪氨酸,未发现NE改变;但如在损伤前给予苯氧苄胺,血压下降32%,病变明显减轻,说明血压较组织NE对脊髓出血更有关。有的研究者报告,尽管在损伤1小时后,损伤处脊髓NE含量增高,但在切除肾上腺的动物反而较正常为低。还有研究者应用放射酶测定,发现脊髓横断后,血浆NE立即升高,但维持时间较短,说明与血流动力学变化有关。脊髓损伤后引起的中央性出血性坏死如非由于脊髓内源性儿茶酚胺增加所致,则很可能使血液循环中的NE增加进入脊髓实质而引起进行性自溶。是否因血浆NE水平升高,改变血-脊髓屏障的渗透性而聚集于损伤的脊髓内还未得到证实。另一种看法认为NE并不引起中央性出血坏死,而是通过α及β受体刺激花生四烯酸的代谢,导致脂质过氧化物和自由基的生成增加使细胞膜受到损害。不同研究者测定的NE、DA及5-HT平均数值如表23-2所示。
表23-2 不同作者测定的NE、DA及5-HT平均数值

二、NO的变化
在中枢神经系统中,一氧化氮(NO)主要由血管内皮细胞、神经细胞和神经胶质细胞的一氧化氮合成酶(NOS)催化L-精胺酸胍基末端氮原子和氧结合而成,具广泛生物活性。NOS是一组同工酶,根据功能可分为结构型(cNOS)和可诱导型(iNOS)。NO是一种带有不成对电子的气体,生物活性非常活泼,半衰期仅为1~5秒。NO具有细胞间第二信使和神经毒性作用,脊髓中NO合成酶阳性神经元主要见于后角浅层、背根神经节中的内脏神经元、中间外侧柱和中央管周围。NO在神经系统信息传导和发育中起作用。已知谷氨酸与NMDAR结合后,能触发阳离子通道的开放,Ca2+进入细胞与钙调蛋白结合,刺激NOS的活性,产生NO。在原代培养的神经元中加入NOS抑制剂,能拮抗NMDA介导的谷氨酸的神经毒性。实验证实,前根撕脱能诱导脊髓前角运动神经元表达NOS,而前根切断则不能。脊髓全切或半切能诱导两侧或切断侧脊髓背侧和神经元表达NOS。上述现象可能是因神经根撕脱后,轴突的SCs鞘不能被保存,失去了神经元主要神经营养因子的来源,而使神经元NOS产生表达,导致NO过量产生而引起神经元死亡。脊髓损伤后,NOS活性增强,NO含量增高,同时脊髓水肿加剧。NO激活可溶性鸟苷酸环化酶,增加细胞中环鸟苷酸(cGMP)的水平,通过调节细胞膜离子通道,引起Na+、Ca2+等分布不平衡,导致细胞水肿。NO还可能有氧自由基作用,造成脂质过氧化,破坏膜结构,而引起血-脑屏障损伤,加剧脑水肿。NO还是一种血管舒张因子。脊髓损伤后,局部NO含量升高,在一定程度上可扩张血管,增加脊髓血流量,改善局部缺血缺氧状态,但它同时又可加重细胞坏死水肿。NO具有双重性能,既是神经细胞和内皮细胞的信息递质,也具有神经保护作用,又有细胞毒性作用,造成神经损伤。这两种不同的作用取决于NO离子的氧化还原状态,氧化型NO具有神经保护作用,还原型NO具细胞毒性作用,其主要表现为:①通过对DNA复制的抑制和对DNA的脱氨基作用,影响靶细胞的修复和蛋白质的合成;②通过与靶细胞的含铁酶和蛋白形成一种NO-铁复合物,如与线粒体电子传递体系中的关键酶相互作用,可抑制酶活性或使其失活;③NO本身是一种活性自由基,与超氧阴离子结合后,更增强其活性,同时使超氧阴离子成为更有力的脂质过氧化激活剂。NO能增加脊髓损伤局部兴奋性氨基酸(EAAs)的释放,其原因是多方面的。脊髓损伤后NO大量释放,加重神经组织损害。NO能抑制Na+-K+-ATP酶的活性,引起神经细胞去极化,促使EAAs的释放。NO本身具有极强的化学活性,作用于失活EAAs的酶,使已释放的EAAs不能迅速降解。NO还可能抑制或破坏EAAs的重摄取功能。所以,NO通过对EAAs的释放、灭活和重摄取多方面作用而使细胞外液EAAs浓度升高。脊髓损伤后,NOS活性升高,又因巨噬细胞等炎性细胞浸润,局部NO产生更多,应用小剂量非特异性NOS竞争性抑制剂L-NAME治疗可减轻组织损害,但大剂量可使组织破坏加重,神经功能不能恢复,说明NO在参与继发性脊髓损伤中,既起保护作用,又起破坏作用。一般认为,由神经细胞、巨噬细胞等产生的NO可以介导兴奋性神经毒性,与氧自由基产生过氧化阴离子HO·,使含铁、硫的酶类失活,破坏DNA双螺旋结构,使组织结构破坏;另一方面,由内皮细胞、平滑肌细胞及血管周围的交感神经产生的NO可舒张血管,增加血流,减少血小板聚集、粘附,避免血栓形成而具有保护作用。正常情况下,NOS与其地底物之间处于饱和状态。组织损伤后,由于血流下降,酶活性增加,使底物减少或对其需要量增加,此时给予合成NO的前体物质L-精氨酸可反应生成NO。对早期脊髓损伤,NO的舒血管功能对神经功能的恢复有一定作用。但由于不可避免的神经源性NO增加,必然影响最终治疗结果。因此,如何控制脊髓损伤后NO的产生对减轻继发性损伤有重要的作用。
三、自由基的变化
自由基是具有异常反应的分子,继发于其外轨道不成对电子,能经链式反应扩展。在氧自由基链式反应累及脂肪酸称为脂质过氧化(lipid peroxidation,LOP)。此反应中生物膜的磷脂及胆固醇成分被自由基反应(free-radical reaction,FR)所破坏。正常细胞代谢途径中氧化还原反应可产生氧自由基有:超氧自由基(O2-)、羟自由基(·HO)及过氧化氢(H2O2),在正常状态下,细胞通过一些天然存在的抗氧化合物管制FR产生的有害作用,如超氧化歧化酶可以清除超氧阴离子,通过催化使其转化为过氧化氢和氧,而过氧化氢酶可使过氧化氢还原为水。FR病理反应可引起LOP并使细胞膜节裂,病理性FR的产生及其有害作用可引起脊髓损伤后缺血。急性脊髓损伤,FR引起LOP表现为过氧化,多不饱和脂肪酸分解产物增多,胆固醇减少伴胆固醇氧化产物、鸟苷酸环化酶的活性及相应cGMP增加;组织内抗氧化物如坏血酸及α-生育酚减少;磷脂依赖性膜结合Na+-K+-ATP酶抑制等。脊髓损伤后,自由基的主要来源为:①缺血缺氧引起的能量代谢障碍,ATP减少,致使线粒体内电子传递速度降低,传递脱节及传递链终端电子不足,结果氧还原不完全而产生超氧阴离子O2-。与此同时,大量辅酶Q(CoQ)及黄素腺嘌呤二核苷酸(FAD)被释放,当有少量氧存在时,二者可自氧化产生超氧自由基:CoQ+O2→CoQ+O2-。②脊髓灰质局部出血,红细胞释放大量氧合血红蛋白和含铁化合物。氧合血红蛋白自氧化高铁血红蛋白过程中产生O2,Fe2+和Cu2+的释放加速O2-的产生。③脊髓损伤后,细胞膜离子泵活性降低,伤区膜通透性增加,促使大量Ca2+内流,Ca2+激活依赖Ca2+的蛋白酶,黄嘌呤脱氢酶转化为黄嘌呤氧化酶,在次黄嘌呤被氧化为尿酸的过程中产生O2-,另外,细胞内游离Ca2+增加,激活多种蛋白酶及磷脂酶A2,发生脂质过氧化,游离脂肪酸释出花生四烯酸,产生大量O2-。④脊髓损伤后释放出大量的儿茶酚胺,在Fe2+存在下,可自氧化产生自由基;或通过受体机制,使细胞内Ca2+及cAMP增加,激活磷脂酶A2,影响细胞膜功能,刺激花生四烯酸代谢而产生O2-。脊髓损伤后的病理性自由基反应可能是脊髓进行性组织坏死的病理改变分子基础之一。损伤后微循环障碍是因损伤后产生的脂质过氧化物抑制前列腺素合成,血小板聚集所致。由于PUFA胆固醇等脂质减少之前,抗氧化剂已下降,说明损伤后首先因自由基产生增加,大量消耗了氧化剂之后,才引起脂质过氧化损害。这种损害使细胞膜上的一些重要依赖于脂质的酶,如Na+-K+-ATP酶失活,膜的通透性改变。另外,溶酶体和线粒体膜的破坏造成细胞内环境紊乱,氧化磷酸化障碍及细胞膜功能丧失。血管内皮细胞的脂质过氧化可使内皮细胞破坏,血管通透性增强,引起水肿和微血管内凝血,加重了缺血、缺氧,造成了广泛坏死。脊髓损伤后血流降低可能由于血管痉挛或微循环阻塞。毛细血管内皮可发生自由基的损害,产生过氧化酶,抑制前列环素的正常形成。前列环素由内皮细胞自花生四烯酸衍生的前列腺素内生过氧化物产生,对主动防止血小板粘连及聚集是需要的。Demopoulos等从分子病因上发现,早在脊髓损伤后1小时,化学上可以看到自由基反应,发展至膜脂、磷脂及胆固醇破坏。酶的生化测定显示中枢神经系统损伤后,膜脂有不同程度的降低,扫描电镜肯定脊髓实质微循环障碍,伤后1小时在毛细血管及其他小血管即有内皮损伤,2小时后变得更为严重,并向远、近侧扩散。这3种变化,即脂质的自由基损害、酶的抑制及微循环障碍均有潜势向损伤部位上下扩展,特别是毛细血管内皮损害,可向近端发展。如果细胞膜的自由基反应能受到控制,血供保持正常,可能会大大减少损害的程度。脊髓损伤后,神经细胞膜、神经胶质、血管等发生一系列不可逆的改变,脊髓退变,血流明显减少,可能是由于血管痉挛或毛细血管栓塞所致。脊髓及中枢神经系统其他部分较其他生命器官对细胞膜的完整性要求更为严格。血-中枢神经系统屏障保持独特的环境,排除某些物质,并增强某些物质如抗坏血酸的运输。脊髓损伤早期的病理改变是缺血,其所引起的继发性损伤与自由基的作用密切相关。缺血、缺氧的器官,在再灌注及再给氧时往往出现更为严重的损伤。离体大鼠心脏实验表明,缺氧3.5~4.0小时后,细胞内肌酸磷酸激酶大量释放,说明缺氧后再给氧更可加重损伤。氧可以还原为O2-等自由基,线粒体呼吸链中的CoQ也可自动氧化,产生O2-。缺血和再灌注损伤可使脂质过氧化反应增强。脂质过氧化反还可以引起多种形式的组织损伤,如增加细胞膜的通透性、降低Ca2+在肌浆网中的转运、改变线粒体功能等。另外,缺血组织中,清除自由基的体系功能较弱也可能是自由基增多的原因。缺氧可以降低细胞中抗自由基氧化的防御体系的作用,再给氧时不能对产生的自由基有效地清除,从而引起组织损伤。细胞含内源性自由基清除剂,如超氧化物歧化酶(SOD)及谷胱甘肽,防止正常细胞呼吸所产生的自由基所致的损伤。但在损伤情况下,形成过多的自由基破坏了细胞防御机制。自由基可攻击脂质膜的多不饱和脂肪酸产生不同的醛,如丙二醛(MDA)和4-hydroxynonenal(HNE)。它们可以破坏蛋白,形成醛-蛋白复合物以改变蛋白质的二级及三级结构。醛与自由基不同,可长期存活,从起始处可弥散攻击远处细胞内、外蛋白质。HNE为醛中最具毒性者,对于脂质过氧化反应较MDA是更特异的指标。Baldwin等对大鼠脊髓T10打击致伤,对脊髓局部外渗血浆应用单克隆抗体测定IgG,并用脂质过氧化反应的醛副产物HNE与蛋白结合以评估氧化应力。动物于伤后1小时至7天处死,伤后2天可以看到HNE蛋白最明显染色,在T10伤区头尾侧呈逐渐不对称反应。尾侧T11及T112水平,HNE蛋白复合物及IgG染色均较头侧T8及T9水平明显增高。对HNE蛋白复合物免疫染色神经元阳性的比率也以尾侧较高。从伤后1小时到2天,HNE蛋白反应产物逐渐增多,说明在此阶段脂质过氧化反应呈上升趋势,伤区头尾段脊髓可发生继发性损伤。HNE蛋白免疫阳性的神经元、轴突、胶质细胞及神经毡可能包括突触均承受氧化应力,功能受到损害而致动物运动丧失。伤后7天,除紧邻损伤区外,HNE蛋白反应产物不出现,说明这些区域自由基产生及脂质过氧化均减轻,细胞内蛋白恢复正常,运动功能有所恢复。伤后7天在紧邻伤区部位,反应产物显示氧化应力的进展。细胞暴露于HNE后可迅速消除谷胱甘肽,引起钙体内平衡紊乱、钾通道功能不良,抑制DNA、RNA的合成,抑制呼吸和糖酵解,Na+-K+-ATP酶和蛋白激酶C活性也降低。由于HNE与膜磷脂相互作用,线粒体膜流动性也受到损害。HNE还可诱导细胞凋亡,是脊髓损伤后细胞死亡的一种机制。HNE浓度极低时对培养的神经元即具有毒性,突出体暴露于HNE可表现为谷氨酸摄入,葡萄糖输送及线粒体功能损害。所以这种醛对突触有破坏作用而引起功能的丧失。IgG染色显示渗出血浆蛋白的定位和暴露于血浆毒素的区域。Noble及Wrathall曾观察到在脊髓横断伤尾侧与等距离近侧相比有一较大的血-脊髓屏障裂口,认为管理血管通透性的较高神经中心白质传导束损害可能起作用,在损伤尾侧也有较大星形胶质细胞反应。Kaptanoglu等将大鼠分为7组,第1、2组仅在第6、7胸椎实行椎板切除,分别于术后立即及术后2小时取材,第3组采用Allen打击法对胸椎给予50mg·cm打击,第4组伤后立即向腹膜腔内注入50mg/kg的琥珀甲基泼尼松龙(MPSS),5、6、7组分别于伤后立即向腹膜腔内注入自由基清除剂Mex 1mg/kg、10mg/kg及50mg/kg。各组动物均于伤后2小时处死取材。应用丙二醛测试脂质过氧化反应示:3、5组明显升高,1、2、4、6及7组无明显差异。超微结构检查示:第3组(单纯脊髓损伤)伤后2小时轴突细胞骨架遭受严重损害,表现为微管和神经丝丧失,在临近轴突的髓鞘内有空区,线粒体肿胀,嵴丢失,白质中轴浆丢失,轴突髓鞘变薄,层次之间可出现裂隙,许多轴突有空泡变性,呈蜂窝状。灰质中多数神经元有水肿,并有膜缺损,细胞质内有许多空泡,无细胞器、细胞核的染色体成丛状。第4组(MPSS组)白质中的神经丝及轴突肿胀,线粒体也肿胀,髓鞘较薄并有裂隙。灰质中的轴突变化同第3组相似,但细胞质变化较轻。第5组(Mex 1mg/kg组)与第3组相似,仅神经丝密度较大,有水肿,线粒体嵴消失。第6组(Mex 10mg/kg组)白质中大部分髓鞘结构及神经丝密度正常,有些髓鞘出现裂隙,有些线粒体嵴也消失,灰质细胞质有轻度水肿,大部分膜完整。第7组(Mex 50mg/kg组)灰、白质超微结构基本正常。由此可见,自由基及其引发的脂质过氧化是导致脊髓损伤后继发性损伤病理改变加重的重要原因,其反应机制大体如图23-2所示。值得指出的是,脊髓损伤后还有其他的功能性物质在起作用,如兴奋性氨基酸含量的增加可导致神经细胞的损伤;甘烷类包括血栓素、白三烯的含量的变化也可使脊髓血流下降,加重损伤;另外,P物质、内皮素、血管内皮生长因子、血小板活化因子、内源性阿片肽、类固醇及钙通道的变化都会影响其损伤后的病理及病生变化。它们的作用不是单一的,联合在一起非常复杂,在此不再详述。
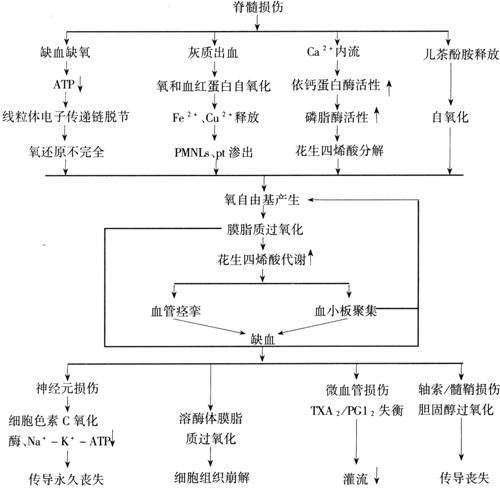
图23-2 脊髓损伤后自由基的来源、过程及作用